
Por Rita Zilhão Faculdade de Ciências de Lisboa – Portugal

O ciclo de vida do virus IAV.
Nature Reviews Microbiology (2011) 9, 590-603
O Vírus da Gripe A (IAV – influenza A virus) apresenta um grande desafio para a saúde pública. Esse vírus infecta as células epiteliais do trato respiratório, causando a sintomatologia gripal por todos nós conhecida e já de alguma forma por nós vivida. Mas, mais grave ainda, é que é um vírus persistente e que tem sido responsável por diversas epidemias (surtos relativamente localizados do ponto de vista geográfico) e algumas pandemias (surtos com uma distribuição geográfica muito mais alargada), e por vezes com um índice considerável de morbidade (relação entre o número de indivíduos sãos e doentes)
Mas que características têm este vírus para que tal aconteça?
Após infectar uma célula, o vírus multiplica-se, produzindo novas cópias do seu material genético (genoma), que serão empacotadas, isto é, envolvidas por um invólucro proteico formando os chamados viriões (vírion no português brasileiro). Essa descendência viral é então libertada indo, por sua vez, infectar novas células epiteliais. Uma característica distinta do IAV é o seu genoma segmentado, constituído por oito moléculas de RNA independentes. O genoma segmentado permite que quando uma célula de um hospedeiro é co-infectada com duas ou mais diferentes estirpes de influenza possa ocorrer, durante o empacotamento, uma combinação diferente dos oito segmentos de RNA. Geram-se novos arranjos do genoma viral e como tal geraram-se novas estirpes IAV que, eventualmente, podem ser mais agressivas. É essa possibilidade de formar novas combinações genômicas que tem permitido ao vírus da gripe ultrapassar as barreiras entre a especificidade das espécies (por exemplo, deixa de haver espécies específicas só de humanos, ou de aves), e que está na origem das grandes pandemias de influenza.
Mas como ocorre esse reconhecimento dos segmentos genômicos entre si, para que só um de cada seja incluído no novo virião? A biologia molecular do rearranjo dos segmentos do genoma do vírus da gripe ainda não está completamente esclarecida, mas provavelmente envolve múltiplos mecanismos que selecionam uma compatibilidade funcional e física dos oito diferentes segmentos dentro de uma nova estirpe. De facto, já se sabe que cada segmento contém sequências específicas que são responsáveis pelo empacotamento de um único conjunto dos oito segmentos de RNA em viriões infecciosos.
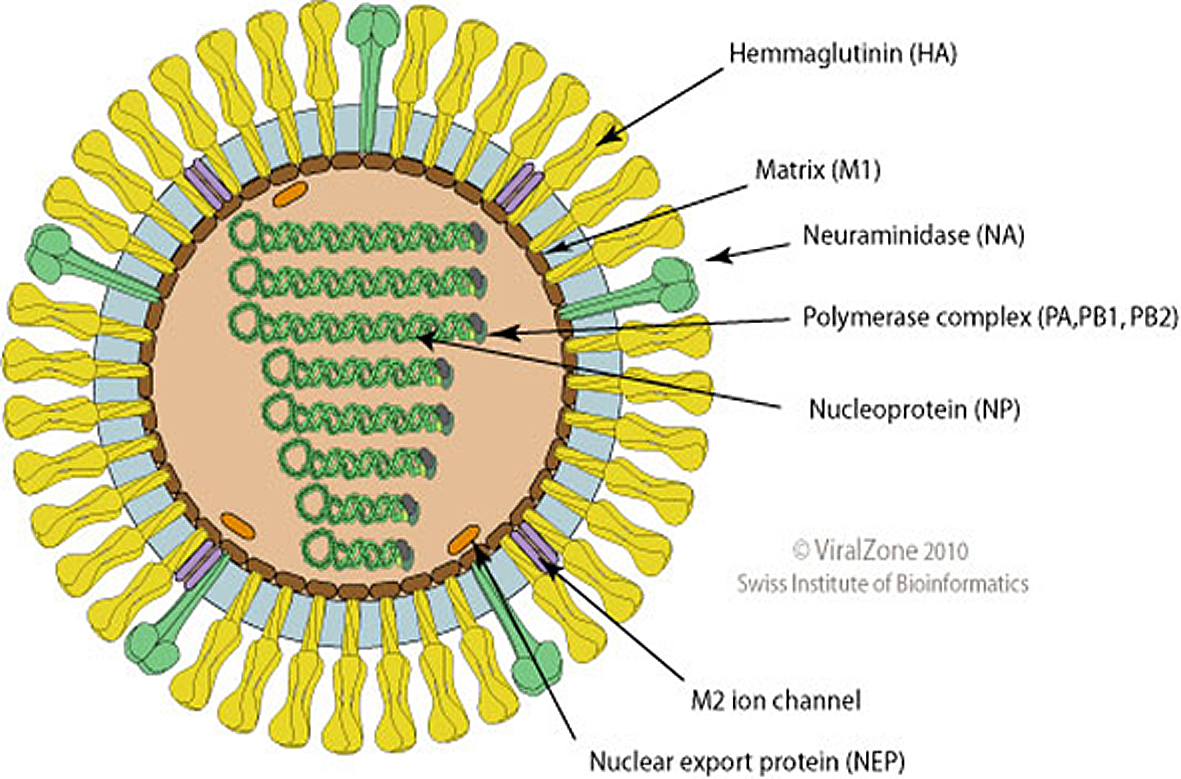
O genoma segmentado do vírus IAV.
http://viralzone.expasy.org/all_by_species/6.html
Por outro lado, também os investigadores têm procurado perceber o que está na base do perfil dos diferentes padrões destes rearranjos genômicos de forma a poder antecipar combinações mais patogênicas. Na verdade, há interesse em entender: 1- Qual a dinâmica populacional dos segmentos nos vírus coinfectantes, incluindo a evolução temporal da população viral. Em outras palavras, saber se há rearranjos mais favorecidos? Qual a frequência dos diferentes rearranjos? Como vão evoluindo os diferentes rearranjos no tempo?; 2- Qual o efeito de cada rearranjo num cenário de fitness (capacidade de se reproduzir e prosperar), incluindo a seleção de mutações que ocorrem nos genomas. Um entendimento claro das associações entre padrões de mutação específicos e rearranjo teria amplas implicações para a saúde pública (prevenção e desenvolvimento de vacinas, por exemplo).
Várias estratégias experimentais e de análise bioinformática têm procurado responder a essas questões. Assim, recentemente, utilizando uma estratégia inovadora in vitro, os cientistas descobriram que a evolução viral provocada pelo rearranjo do genoma segmentado de IAV pode ser acelerada e potencialmente ativada pelo aparecimento de um pequeno número de mutações individuais.
Como os cientistas procederam para chegar a esta conclusão? Monitoraram a evolução dos rearranjos de todos os genomas virais que resultaram de experiências e coinfeção (infeção com duas estirpes diferentes de IAV). Através da determinação da sequência nucleotídica desses genomas e utilizando métodos bioinformáticos e matemáticos quantificaram a abundância relativa das frequências dos segmentos das duas estirpes parentais de vírus influenza ao longo do tempo. Também mediram os coeficientes de seleção dos segmentos rearranjados, ou seja, inferiram uma evolução temporal dos rearranjos genômicos.
Curiosamente, observaram várias mutações pontuais nos genomas provenientes de coinfeção, mutações essas que não surgiam quando se infectava com os vírus parentais puros individualmente, ou seja, quando não havia a possibilidade de ocorrer um rearranjo entre genomas de origens diferentes.
Assim, os dois fatores que conduzem aos efeitos que as novas estirpes manifestam, e que estão na origem da rápida adaptação e evolução do vírus da gripe são: a natureza segmentada do genoma viral de influenza, que facilita o rearranjo dos segmentos, mas também a seleção de diferentes mutações que é provocada por combinações genéticas específicas dos segmentos. Para os mais entendidos, esse fenômeno está muito próximo da noção de epistasia (isto é, uma situação na qual a expressão de um gene depende da ação de outro gene que não seja um de seus alelos).
Para saber mais, acesse o artigo original, clicando aqui.

Republicou isso em maismaismedicina.